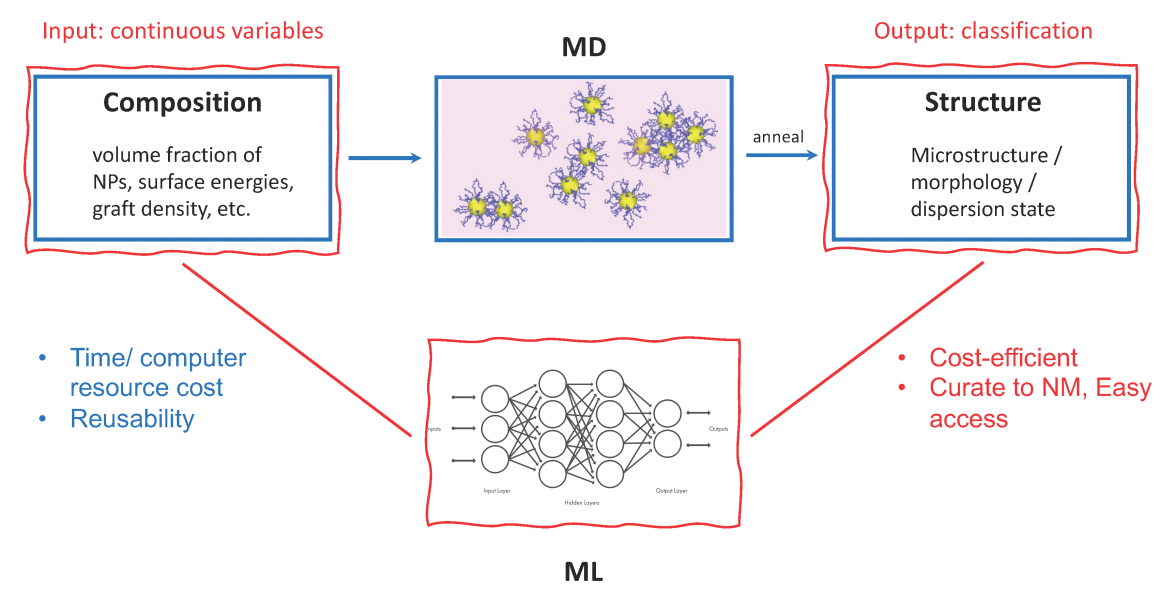
A major challenge in design of nanostructured polymers and polymer nanocomposites (PNCs) is the nearly infinite design space due to the enormous range of materials selection, surface chemistries, and processing methods. Molecular modeling techniques such as Molecular Dynamics (MD) simulations can reveal the microstructure of PNCs by simulating the system undergoing an annealing process analogous to the synthesis step in experiments. However, molecular modeling also faces the problems of an overly large design space. For instance, there is a large range of compositional parameters that play a critical role in determining the microstructure of PNCs, including matrix and filler selection, filler surface treatment, and filler volume fraction, each of which has many nuanced choices which can significantly impact material response. Exploring the entire range of these parameters is not practical experimentally or computationally. With the recent developments in Artificial Intelligence, a predictive model that helps understand the composition—structure relationship of PNCs has the potential to make significant strides to address the existing limitations. In this work, we perform MD simulations of PNCs within a carefully defined range of compositional parameters that sufficiently spans a region of the space. We then characterize the dispersion state of fillers from the MD simulation trajectories, and build interpretable machine learning models to shed light on the composition—structure relationships. The outcome will be a robust metamodel which will enable intelligent interpolation in the design space without full direct numerical simulation or experimentation and will help guide material understanding and design.